Daily magnesium fluxes regulate cellular timekeeping and energy balance
Kevin A. Feeney1, Louise L. Hansen2, Marrit Putker1, Consuelo Olivares-Yañez3, Jason Day4, Lorna J. Eades5, Luis F. Larrondo3, Nathaniel P. Hoyle1, John S. O'Neill1,#, and Gerben van Ooijen2,#
1MRC Laboratory for Molecular Biology, Francis Crick Avenue, Cambridge Biomedical Campus, Cambridge CB2 0QH, UK2School of Biological Sciences, University of Edinburgh, Max Born Crescent, Edinburgh EH9 3BF, UK3Millennium Nucleus for Fungal Integrative and Synthetic Biology, Departamento de Genética Molecular y Microbiología, Facultad de Ciencias Biológicas, Pontificia Universidad Católica de Chile, Casilla 114-D, Santiago, Chile4Department of Earth Sciences, University of Cambridge, Downing St, Cambridge CB2 3EQ, UK5School of Chemistry, University of Edinburgh, David Brewster Road, Edinburgh EH9 3FJ, UK
Abstract
Circadian clocks are fundamental to the biology of most eukaryotes, coordinating behavior and physiology to resonate with the environmental cycle of day and night through complex networks of clock-controlled genes1–3. A fundamental knowledge gap exists however, between circadian gene expression cycles and the biochemical mechanisms that ultimately facilitate circadian regulation of cell biology4,5. Here we report circadian rhythms in the intracellular concentration of magnesium ions, [Mg2+]i, which act as a cell-autonomous timekeeping component to determine key clock properties in both a human cell line and a unicellular alga that diverged from metazoans more than 1 billion years ago6. Given the essential role of Mg2+ as a cofactor for ATP, a functional consequence of [Mg2+]i oscillations is dynamic regulation of cellular energy expenditure over the daily cycle. Mechanistically, we find that these rhythms provide bilateral feedback linking rhythmic metabolism to clock-controlled gene expression. The global regulation of nucleotide triphosphate turnover by intracellular Mg2+ availability has potential to impact upon many of the cell’s >600 MgATP-dependent enzymes7 and every cellular system where MgNTP hydrolysis becomes rate limiting. Indeed, we find that circadian control of translation by mTOR8 is regulated through [Mg2+]i oscillations. It will now be important to identify which additional biological processes are subject to this form of regulation in tissues of multicellular organisms such as plants and humans, in the context of health and disease.
Circadian rhythms occur cell-autonomously and are not restricted to metazoans or multicellular organisms, being found throughout eukaryotes and some prokaryotes9. Although explicit clock gene identities share no similarity across phylogenetic kingdoms, in every case temporal orchestration of gene expression is driven by timekeeping mechanisms that result in rhythmic clock protein transcription factor activity. In human cells, for example, a heterodimeric complex of BMAL1 and CLOCK positively regulates the expression of genes (Period1/2, Cryptochrome1/2) that encode its own repressor complex9, whereas in the marine unicellular alga Ostreococcus tauri, a reduced version of the stereotypical plant-like circadian clock consists of a feedback loop between the morning-expressed MYB-like transcription factor CCA1 and the evening-expressed protein TOC110. Intriguingly, the role of enzymes such as casein kinase 1 and 2 in the post-translational regulation of clock protein activity/stability, and the speed at which biological clocks run, is functionally conserved across eukaryotes11. Also, we recently reported a circadian rhythm in the redox state of peroxiredoxin proteins that is conserved across phylogenetic kingdoms4,12. Critically, this metabolic rhythm persists in the absence of nascent gene expression, both in human cells (anucleate erythrocytes)13 and in Ostreococcus, which ceases mRNA production upon prolonged photosynthetic inactivity under constant darkness14, indicating that circadian regulation of cellular metabolism is not strictly reliant upon rhythmic transcription. These and a number of other observations render it plausible that circadian rhythms observed in diverse eukaryotes incorporate features of a post-translational timing mechanism that was present in the last eukaryotic common ancestor, LECA15.
A rich diversity of evolutionarily conserved membrane transporters and channels mediate uptake of ions and micronutrients essential for cellular biochemistry, with several studies having reported their circadian regulation in a variety of contexts (e.g.16), and with membrane models of the circadian clock actually predating the identification of any clock genes17,18. It is plausible that rhythmic regulation of ion transport may have conferred an adaptive advantage upon early eukaryotes, allowing the global regulation of biochemical equilibria and reaction rates to tune cellular metabolism with environmental cycles. We therefore asked whether circadian regulation of transmembrane ion transport might constitute a fundamental feature of circadian timekeeping in eukaryotic cells. To find evidence for such regulation in modern organisms, we compared two eukaryotic lineages separated by ~1.5 billion years of evolution6: human U2OS cells and Ostreococcus tauri.
Inductively Coupled Plasma Mass Spectrometry (ICP-MS) was employed to generate an unbiased analysis of the total cellular elemental composition, including all organelles and cellular structures19,20, over circadian time series. In Ostreococcus, clear daily rhythms were detected under natural light/dark cycles for many different ions, including potassium and magnesium (Fig. 1a and Extended Data Fig. 1). Analyses of cells maintained under constant light revealed that, whilst rhythms of some metal ions ceased under these conditions, oscillations in potassium and magnesium persisted, indicating their regulation by cell-autonomous circadian clock mechanisms. Strikingly, circadian rhythms of magnesium and potassium were also observed in non-proliferating human U2OS cells maintained over three days under constant conditions (Fig. 1b and Extended Data Fig. 2). The oscillation in intracellular potassium is likely to be a consequence of rhythmic Na+-dependent pump activity, as circadian regulation of sodium-dependent solute-transport and plasma membrane ATPases has been reported widely (e.g.2,16,21,22). Calcium is largely found in, and released from, intracellular stores and although calcium has a clearly established role in circadian rhythms11, we infer the absence of any obvious cell-autonomous Ca2+ oscillations to mean that its net cellular flux does not vary over the circadian cycle in these cells. We considered the oscillation in Mg2+ to be of particular interest, since Mg2+ is an essential cofactor for (deoxy-) nucleotide triphosphates. Cellular Mg2+ therefore has the potential to regulate many intracellular metabolic reactions, through its requirement for the activities of >600 enzymes7, including those involved in ATP production, as well as DNA, RNA and protein synthesis. Furthermore, free intracellular Mg2+ can act as a second messenger. For example, epidermal growth factor stimulation induces a rapid increase in [Mg2+]i, which acts via highly Mg-sensitive mTOR to activate protein synthesis without any change in total ATP levels23.
Firstly, we exploited the role of Mg2+ as an ATP cofactor, to measure freely available intracellular magnesium concentrations in cellular extracts using the MgATP-dependent enzyme firefly luciferase (Fig. 1c,d). Our initial ICP-MS observations were confirmed using this assay, and we observed clear rhythms of [Mg2+]i over two days under constant conditions in both cell types. Bioluminescent reporters for clock gene activity, recorded in parallel, confirmed the [Mg2+]i oscillation to occur roughly in antiphase with circadian markers normally expressed around (subjective) dawn (CCA1-LUC, Per2:luc, Fig. 1a,b). Conservation between human and algal cells indicates that rhythmic cation transport might constitute a general feature of cellular rhythmicity. We therefore investigated whether such oscillations were also present in the fungus Neurospora crassa, representing the third eukaryotic kingdom. Similar to our observations in algal and human cells, a circadian rhythm in cellular magnesium content was observed in antiphase with the abundance of the clock protein FRQ9 (Extended Data Fig. 3a-c). We also observed cellular magnesium rhythms in cultured mouse fibroblasts isolated from adult lung, indicating that magnesium rhythms are also present in non-transformed, terminally differentiated mammalian cells (Extended Data Fig. 3d). These rhythms were disrupted in fibroblasts isolated from Cryptochrome1-/-,Cryptochrome2-/- mice, suggesting their dependence upon clock gene activity. Incidentally, we note that approximately 24 h [Mg2+]i rhythms occur cell-autonomously, are temperature-compensated (Extended Data Fig. 4) and entrain to relevant external cues and are therefore circadian by definition24.
The rhythm in total cellular Mg2+ measured by ICP-MS must result from daily cycles between net cellular Mg2+ influx and efflux, through circadian regulation of plasma membrane Mg2+ channel and transporter activity7. All known magnesium transporting proteins in animals (channels TRPM7, MAGT1, MMGT1 and CNMM3, as well as Mg2+-transporter SLC41) exhibit circadian rhythms at the mRNA level in four or more tissues25 (Extended Data Fig. 5). Ostreococcus encodes homologs of TRPM7, CNMM3 and SLC41, which are also differentially expressed over the daily cycle (Extended Data Fig. 5).
Moreover, siRNA-mediated knockdown of each Mg2+-channel/transporter in U2OS cells results in lengthened circadian period26, suggesting that as well as being clock-regulated, [Mg2+]i might also feed back to regulate the cellular clock.
To determine whether [Mg2+]i oscillations are relevant to timekeeping mechanism therefore, we next employed inhibitors of magnesium transport. Cobalt(III)hexammine (Co(NH3)62+, CHA) and cobalt(III)chloro-pentammine (Co(NH3)5Cl2+, CPA) closely resemble a single-solvation shell hydrated Mg2+ ion, and have been shown to block Mg2+ transport through at least two different transporters/channels27,28. We found both compounds to dose-dependently increase [Mg2+]i in both cell types (Fig. 2a,b and Extended Data Fig. 6), indicating that these compounds do act to block Mg2+ transport. Increased [Mg2+]i was associated with clear dose-dependent lengthening of circadian period (Fig. 2c-f). Importantly, the effects of CHA on circadian period were dependent on the concentration of extracellular magnesium (Extended Data Fig. 7a-d), indicating a specific role for magnesium in determining the speed at which both algal and human cellular clocks run. To further substantiate this observation, we used quinidine, an inhibitor that acts on several ion transport activities including the SLC41 Na+/Mg2+ antiporter29. Similarly to CHA and CPA, quinidine led to dose-dependent accumulation of intracellular Mg2+ and lengthening of circadian period in both cell types (Fig. 2). SLC41 constitutes the sole protein known to exhibit sodium-dependent Mg2+-transport activity29 that is conserved between human and Ostreococcus cells and so, to test its specific cellular clock function, we performed siRNA-mediated SLC41 knock-down: observing a clear Mg2+-dependent lengthening of circadian period (Extended Data Fig. 7e-g).
We also observed that depletion of magnesium from the growth medium led to reduced [Mg2+]i, and had dramatic effects on the amplitude and period of the circadian clock in Ostreococcus (Fig. 3a,b). Although prolonged growth in low Mg2+ media had adverse effects on cell viability of the U2OS line, cells that were simply transferred to Mg2+-free media showed reduced [Mg2+]i and exhibited circadian rhythms with increased period and decreased bioluminescence amplitude relative to normal media controls (Fig. 3c,d). In neither case was the effect of [Mg2+]i-depletion attributable to decreased ATP availability, since in both cases cellular ATP levels were significantly increased (Fig. 3b,d).
Thus, as observed previously for cAMP signalling30, treatments which constitutively elevate or reduce [Mg2+]i both result in period lengthening of clock gene expression in these cell types, indicating that dynamic circadian regulation of [Mg2+]i might be a cellular clock component. On the other hand however, it remained possible that Mg2+ transport might not contribute to clock mechanism, but instead simply be permissive for cellular timekeeping, analogous to the function of ‘housekeeping’ genes. To distinguish between these two possibilities, we determined whether an enforced transition in [Mg2+]i acts as a state variable for cellular circadian oscillations. Upon introduction of magnesium to Ostreococcus cells starved of magnesium, we observed strict resetting of the subsequent rhythm to subjective dawn, regardless of prior circadian phase, indicating that changes in [Mg2+]i can act as a strong zeitgeber (Extended Data Fig. 8). Therefore, our data indicate that not only does [Mg2+]i exhibit a bona fide circadian rhythm across diverse eukaryotic cells, but also that appropriate manipulation of [Mg2+]i is sufficient to determine the key properties of the oscillation (period, amplitude, and phase), making [Mg2+]i indistinguishable from a core clock component.
We considered that the increased cellular ATP levels we observed during Mg2+ depletion might be attributable to differential sensitivity of MgATP-dependent cytosolic enzymes compared with the organellar ATP synthesis machinery. For example, ATP accumulation was accompanied by a marked reduction in extracellular lactate accumulation in U2OS cultures (Extended Data Fig. 9a), indicative of reduced glycolysis. We therefore considered whether rate-changes in gross cytosolic energy metabolism might be a functional consequence of cell-autonomous circadian [Mg2+]i oscillations. A clear prediction would be that global rates of translation should be limited at circadian phases of low [Mg2+]i, since protein synthesis is one of the most energetically expensive processes that cells undertake.
We assayed translation rate by puromycin incorporation8 in both cell types just before (anticipated) biological dusk and dawn; at the phase of lowest and highest [Mg2+]i, respectively. The Ostreococcus experiment was performed under its natural light/dark cycle so as to best model an organism in its natural environment, whereas the U2OS experiment was performed under constant conditions to model innate peripheral cellular clock function. We observed that both Ostreococcus and U2OS cells did exhibit significantly higher translation rates at the peak of [Mg2+]i, as predicted (Fig. 4c,d). In mammalian cells, the highly MgATP-sensitive mTOR pathway was recently shown to mediate circadian control of translation8. We hypothesised that [Mg2+]i oscillations might act via mTOR to effect circadian translational regulation, and tested this using two pharmacologically distinct mTOR inhibitors (torin1 and rapamycin). Both inhibitors lengthened period dose-dependently, and abolished any additional period lengthening due to depletion of extracellular magnesium that was observed in controls. This clear ‘ceiling effect’ strongly suggests changes in [Mg2+]i act through mTOR activity to regulate cellular circadian period (Extended Data Fig. 9b-e). To test the extent to which marked differences in translation rates between dawn and dusk were attributable to cell-autonomous [Mg2+]i oscillations, we incubated cells acutely with CHA to block magnesium transport before assaying overall translational rates. CHA significantly attenuated the difference in [Mg2+]i between dawn and dusk (Fig. 4a), and also phase-dependently affected ATP levels (Fig. 4b). Crucially, differential translation rates were attenuated similarly by CHA treatment (Fig. 4c,d), indicating that in both cell types, circadian regulation of magnesium levels contributes to circadian rhythms in global translation rate.
Our data support a model (Fig. 4e, Extended Data Fig. 10a) where the cellular clockwork regulates the expression of plasma membrane Mg2+ channels and transporters to generate rhythmic magnesium fluxes. These rhythms appear to facilitate the higher energetic demands and protein production of human cells during the (biological) day, as well as the global down-regulation of ATP turnover and translation in photosynthetic cells at night. Reciprocally, the [Mg2+]i rhythm feeds back to regulate the period, phase and amplitude of clock gene expression rhythms, acting as a “meta-regulator” to integrate metabolic rhythms with transcriptional feedback models of cellular timekeeping. It is noteworthy however, that the magnesium rhythm observed in Ostreococcus persists under transcriptionally inactive conditions14 of constant darkness (Extended Data Fig. 10b,c), in the absence of transcriptional regulation of membrane transport. It is therefore likely that some aspects of circadian magnesium flux are regulated by, and may contribute to, the same uncharacterised non-transcriptional clock mechanism13,14 that also drive persistent peroxiredoxin overoxidation rhythms in transcriptionally inactive cells across taxa, and which we speculate was present in the LECA.
Cell-autonomous rhythms in [Mg2+]i availability have the potential to impart circadian regulation to any cellular system where MgNTP hydrolysis becomes rate limiting. Although the clinical relevance of [Mg2+]i in various tissues is beginning to garner more attention, the interactions between magnesium transport and human health are poorly understood. Further investigation of the downstream consequences of circadian regulation of [Mg2+]i will therefore be important.
There is an ongoing debate about the degree to which T2DM and, more recently, T1DM contribute to AD pathogenesis. This concept has been fueled by the rising prevalence rates of obesity, T2DM, and AD over the past several decades. Moreover, an interrelationship among these entities is suggested by (1) increased risk of developing mild cognitive impairment (MCI), dementia, or AD in individuals with T2DM47,48 or obesity/dyslipidemic disorders;49 (2) progressive brain insulin resistance and insulin deficiency in AD;5,10,26,27 (3) cognitive impairment in experimental animal models of T2DM and/or obesity;50,51 (4) AD-type neurodegeneration and cognitive impairment in experimentally induced brain insulin resistance and insulin deficiency;29,52–55 (5) improved cognitive performance in experimental models and humans with AD or MCI after treatment with insulin sensitizer agents or intranasal insulin;28,56–62 and (6) shared molecular, biochemical, and mechanistic abnormalities in T2DM and AD.47,63–67 The urgency of this problem is spotlighted by the estimated 24 million people in the world with dementia and the expectation that, if current trends continue,68 prevalence rates of AD are likely to double every 20 years in the future. While aging is clearly the strongest risk factor for AD, emerging data suggest that T2DM and dyslipidemic states can contribute substantially to the pathogenesis of AD either directly or as cofactors.68
Epidemiologic studies provide convincing evidence for a significant association between T2DM and MCI or dementia and furthermore suggest that T2DM is a significant risk factor for developing AD.47,69–73 However, those findings are not without controversy,74 and in a longitudinal survey, investigators found that although borderline diabetics had a significantly increased risk for future development of diabetes, dementia, or AD, the risk effects were independent rather than linked.75 What this means is that insulin resistance, i.e., impaired ability to respond to insulin stimulation, can vary among target organs and be present in just one or two organs and not in others, a phenomenon that could explain the lack of complete overlap between T2DM and AD. Correspondingly, the finding that obesity (body mass index [BMI] > 30) without T2DM produces a three fold increase in risk for subsequently developing AD whereas overweight, but nonobese, subjects (BMI 25–30) experience a two-fold increase in risk for AD76 calls into question the specific effects of obesity and T2DM versus a yet unknown associated factor in relation to AD pathogenesis.
Mechanistically, the increased risk of dementia in T2DM and obesity could be linked to chronic hyperglycemia, peripheral insulin resistance, oxidative stress, accumulation of advanced glycation end products, increased production of pro-inflammatory cytokines, and/orcerebral microvascular disease.73 The potential role of cerebral microvascular disease as a complicating, initiating, or accelerating component of AD has been recognized for years.77 However, a magnetic resonance imaging study demonstrated that older adults with T2DM have a moderately increased risk for developing lacunes and hippocampal atrophy and that the severity of those lesions increases with the duration and progression of T2DM.78 Another study showed that T2DM and impaired fasting glucose occur significantly more frequently in AD than in non-AD controls.79
However, since diffuse and neuritic plaques were similarly abundant in T2DM and control brains, and since neurofibrillary tangles, one of the hallmarks and correlates of dementia in AD, were not increased in T2DM,79 the results suggest that T2DM can enhance progression but may not be sufficient to cause AD. Therefore, what remains unclear is the net contribution of T2DM or obesity to the pathogenesis of AD-type neurodegeneration. To address this question, we utilized an established experimental model of chronic high-fat diet (HFD) feeding of C57BL/6 mice to examine the degree to which obesity/T2DM was sufficient to produce histopathological, molecular, and/or biochemical brain abnormalities of AD-type neurodegeneration, i.e., T3DM.
High-fat diet feeding for 16 weeks doubled mean body weight, caused T2DM, and marginally reduced mean brain weight.80 Those effects were associated with significantly increased levels of tau, IGF-1 receptor, IRS-1, IRS-4, ubiquitin, glial fibrillary acidic protein (GFAP), and 4-hydroxynonenal and decreased expression of β actin. Importantly, HFD feeding also caused brain insulin resistance manifested by reduced top-level (Bmax) insulin receptor binding and modestly increased brain insulin gene expression. However, HFD fed mouse brains did not exhibit AD histopathology or increases in APP-Aβ or phospho-tau, nor were there impairments in IGF signaling, which typically occurs in AD.10 In essence, although the chronic obesity with T2DM model exhibited mild brain atrophy with insulin resistance, oxidative stress, and cytoskeleton degradation, the effects were modest compared with AD5,10 and other more robust experimental models of T3DM,28,29 and most of the molecular, biochemical, and histopathological features that typify AD were not present. Therefore, T2DM and obesity may contribute to, i.e., serve as cofactors of AD but by themselves are probably not sufficient to cause AD. Moreover, the findings in the T2DM/obesity model indicate the unlikelihood that brain insulin resistance is sufficient to cause AD and that additional significant abnormalities, such as ongoing DNA damage and mitochondrial dysfunction, are required.
Potential Roles of Obesity and Type 2 Diabetes Mellitus in Alzheimer’s Disease Pathogenesis
Alzheimer’s Disease is Type 3 Diabetes: Evidence from Human Studies
This hypothesis was directly investigated by first examining postmortem cases of advanced AD and determining if the neurodegeneration was associated with significant abnormalities in the expression of genes encoding insulin, IGF-1, and IGF-2 peptides, their receptors, and downstream signaling mechanisms.5 In that study, we demonstrated advanced AD to be associated with strikingly reduced levels of insulin and IGF-1 polypeptide and receptor genes in the brain (Figure 1).
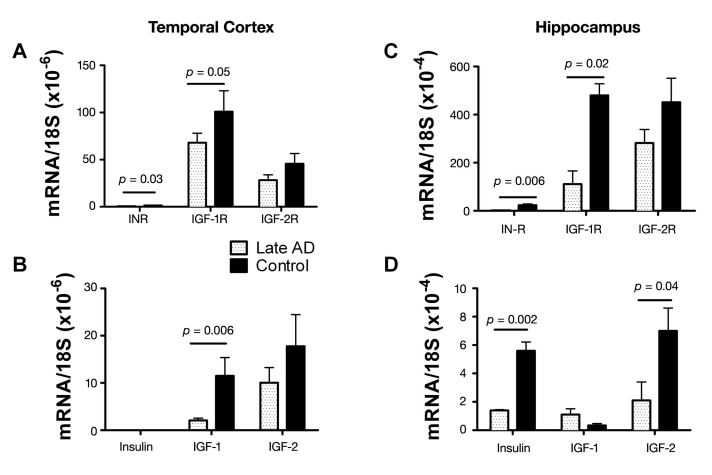
Figure 1. Impaired insulin and IGF (A, C) receptor and (B, D) polypeptide gene expression in late/end-stage AD (A, B) temporal cortex and (C, D) hippocampus.5 Gene expression was measured by qRT-PCR using RNA isolated from the temporal cortex or hippocampus from postmortem histopathologically confirmed cases of severe AD or normal aging. We reverse transcribed mRNA, and the resulting cDNA was PCR amplified. The products were detected continuously with a BIO-RAD iCycler Multi-Color RealTime PCR Detection System. Gene expression was normalized to 18S rRNA measured in the same samples. Graphs depict the mean ± standard deviation of results obtained from 28 AD and 26 control cases. Data were analyzed using Student’s t-tests. Significant p-values are indicated over the bars. Note that insulin gene expression was not detected in temporal cortex.
In addition, all the signaling pathways that mediate insulin and IGF-1-stimulated neuronal survival, tau expression, energy metabolism, and mitochondrial function were perturbed in AD. This study carries additional significance because it established that, like all other pancreatic and intestinal polypeptide genes, the insulin gene was also expressed in the adult human brain. Moreover, the results taught us that endogenous brain deficiencies in insulin, IGF-1, IGF-2, and their corresponding receptors, in the absence of T2DM or obesity, could be linked to the most common form of dementia-associated neurodegeneration in the Western hemisphere. Since the abnormalities identified in the brain were quite similar to the effects of T1DM or T2DM (though none of the patients had either of these diseases), including abnormalities in IGFs,81–83 which are important for islet cell function,84,85 we proposed the concept that AD may represent a brain-specific form of diabetes mellitus and coined the term “type 3 diabetes.” Even before the initial study had been published, it was realized that if brain insulin/IGF resistance and insulin/IGF deficiency were causal in the pathogenesis of AD, the related abnormalities should be detectable in the early stages of disease and possibly worsen as disease progresses. The investigations were extended to examine the brains of patients with different degrees, i.e., Braak stages,86,87 of AD.10 In that study, we measured the expression of genes encoding insulin, IGF-1, IGF-2 polypeptides, and their corresponding receptors as well as tau and amyloid precursor protein (APP). In addition, we used competitive equilibrium and saturation binding assays to further characterize the degree to which growth factor-transmitted signaling was impaired in the brains with different severities of AD. Finally, the study included the measurement of steady-state levels of adenosine triphosphate and genes regulating acetylcholine homeostasis and energy metabolism. Using the previously mentioned approaches, we demonstrated progressive AD Braak stage-dependent reductions in insulin, IGF-1, and IGF-2 receptor expression, with more pronounced deterioration in insulin and IGF-1 compared with IGF-2 receptors, and the lowest levels of gene expression in brains with AD Braak Stage 6 (Figure 2). Therefore, loss of insulin and IGF-1 receptorbearing neurons begins early and progresses with disease such that, in the advanced stages, the deficits are severe and global. These results provided further evidence that the abnormalities in AD are not restricted to insulin signaling pathways, as they also involve IGF-1 and IGF-2 stimulated mechanisms. Analysis of growthfactor polypeptide genes also revealed AD Braak stagedependent impairments in insulin, IGF-1, and IGF-2 polypeptide expression, corresponding with progressive trophic factor withdrawal (Figure 2). Again, the results support the hypothesis that abnormalities in insulin and IGF signaling mechanisms begin early in the course of AD and are therefore likely have an important role in its pathogenesis.
Figure 2. Brain insulin and IGF deficiency and resistance increase with progression of AD.10 Postmortem histopathological studies categorized the brains as having normal aging (Braak 0–1), or mild (Braak 2–3), moderate to severe (Braak 4–5), or end-stage (Braak 6) AD. We used mRNA isolated from fresh frozen frontal lobe tissue to measure insulin, IGF-1, or IGF-2 (A) polypeptide or (B) receptor gene expression by qRT-PCR. Results were normalized to 18S rRNA measured in the same samples. (C) For the competitive equilibrium binding assays, frontal lobe membrane protein extracts were incubated with [125I]- labeled insulin, IGF-1, or IGF-2 in the presence or absence of excess cold ligand. Radioactivity present in membrane protein precipitates was measured in a gamma counter. Specific binding (fmol/mg) was calculated using the GraphPad Prism 4 software. All graphs depict the mean ± standard deviation of results obtained from 9–12 cases per group. Intergroup comparisons were made using analysis of variance (ANOVA) with post hoc Tukey–Kramer significance tests. Significant p-values are indicated over the bars. Note axis break in Panel B.
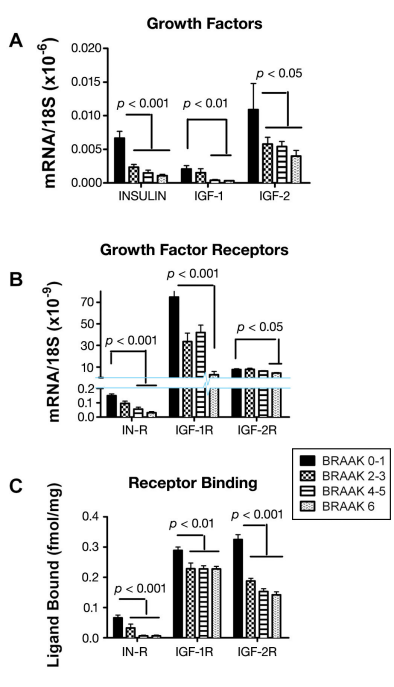
The eventual paucity of local growth-factor gene expression could substantially impair growth-factor signaling and produce a state of growth-factor withdrawal,which is a well-established mechanism of neuronal death. Therefore, to complement the molecular data, we performed competitive equilibrium and saturation binding assays to determine if reduced levels of growth factor receptor expression were associated with and perhaps mediated by impaired ligand-receptor binding as occurs with insulin/IGF resistance. Those investigations demonstrated progressive declines in equilibrium (Figure 2) and top-level binding (Bmax) to the insulin, IGF-1, and IGF-2 receptors but either unchanged or increased binding affinity, suggesting that impaired insulin/IGF actions in AD brains were mediated by decreased polypeptide and receptor gene expression due to cell loss.
Through a series of in vitro and in vivo experiments performed by several groups, including our own, we have been able to draw the conclusion that neuronal and oligodendroglial cell survival and function are integrally related to the integrity of insulin and IGF signaling mechanisms in the brain.10,28,29,31,33,88,89 Similarly, impairments in insulin/IGF signaling lead to deficits in energy metabolism with attendant increased oxidative stress, mitochondrial dysfunction, proinflammatory cytokine activation, and APP expression.4,10,28,89 Correspondingly, the reduced expression of neuronal and oligodendroglial specific genes and the increased expression of astrocytic and microglial inflammatory genes in AD were attributed to progressive brain insulin/IGF deficiency and resistance. Although this point requires the generation of experimental models to demonstrate proof of principle, the finding that microglial, astrocytic, and APP mRNA levels are all increased in the early stages of neurodegeneration supports the inflammatory hypothesis of AD.6 Previous studies demonstrated that microglial activation promotes APP-Aβ accumulation90–92 and that APP gene expression and cleavage increase with oxidative stress.93 Therefore, the mechanism we propose is that impaired insulin/ IGF signaling leads to increased oxidative stress and mitochondrial dysfunction,32,94,95 which induces APP gene expression and cleavage.93 The attendant APP-Aβ accumulations cause local neurotoxicity96–98 and further increase in oxidative stress-induced APP expression and APP-Aβ deposition.
A critical goal in these investigations was to draw connections between brain insulin/IGF deficiency and resistance and the major dementia-associated structural and biochemical abnormalities in AD. In this regard, the postmortem studies demonstrated that the Braak stage-associated declines in tau mRNA paralleled the progressive reductions in insulin and IGF1 receptor expression in AD. In addition, the studies demonstrated AD Braak stage-associated declines in
choline acetyltransferase (ChAT) expression with reduced colocalization of ChAT with insulin or IGF-1 receptor immunoreactivity in cortical neurons. These results correspond with experimental data demonstrating that neuronal tau and ChAT gene expression are regulated by IGF-1 and insulin stimulation.88 Therefore, brain insulin and IGF deficiency and resistance could account for the cytoskeletal collapse, neurite retraction, synaptic disconnection, loss of neuronal plasticity, and deficiencies in acetylcholine production, all of which correlate with cognitive decline and dementia in AD. Altogether, the studies utilizing postmortem human brain tissue provide solid evidence that AD is associated with fundamental abnormalities in insulin/IGF signaling mechanisms that are highly correlated with development and progression of structural, molecular, and biochemical lesions that correlate with dementia. Although the abnormalities noted in AD share features in common with T1MD and T2MD, they are nonetheless distinguished by the dual presence of trophic factor deficiencies and trophic factor receptor resistance, ergo the term “type 3 diabetes.”
Alzheimer’s Disease Is Type 3 Diabetes: Experimental Animal Model Results
The human postmortem brain studies linked many of the characteristic molecular and pathological features of AD to the reduced expression of the insulin and IGF genes and their corresponding receptors. However, without direct experimentation that generates cause–effect data, conclusions drawn from human studies would remain correlative rather than mechanistic. Consequently, we utilized experimental models to demonstrate that diabetes mellitus-type molecular and biochemical abnormalities could be produced in CNS neurons and brain by exposure to streptozotocin (STZ). Streptozotocin is 2-Deoxy-2{[methyl-nitrosoamino)carbonyl]amino}Dglucopyranose, i.e., a nitrosamide methylnitrosourea linked to the C2 position of D glucose. Once metabolized, the N nitrosoureido is liberated and causes DNA damage through generation of reactive oxygen species such as superoxide, hydrogen peroxide, and nitric oxide.99,100 Streptozotocin causes diabetes because it is taken up by insulin-producing cells, such as beta cells, in pancreatic islets. We treated rats with a single intracerebral injection of STZ (ic-STZ) and allowed the rats to grow older for 2 to 8 weeks. The rats were subjected to Morris water maze tests of spatial learning and memory, and their brains were examined for histopathological, biochemical, and molecular indices of AD-type neurodegeneration.
Although a similar model had been generated much earlier by other investigators,101–104 and it was noted that the ic-STZ treatments reduced cerebral glucose utilization104 and oxidative metabolism,101 it inhibited insulin receptor function,95 and it caused progressive deficits in learning, memory, cognitive behavior, and cerebral energy balance,94,103 efforts were not made to connect these effects of ic-STZ to AD by characterizing the neuropathology, molecular pathology, abnormalities in genes expression pertinent to insulin and IGF-1 signaling in brain or by evaluating the integrity of the pancreas. Our goal in generating the model was to demonstrate that AD-type neurodegeneration with features of T3DM could be produced in the absence of either T1DM or T2DM.
The ic-STZ-injected rats did not have elevated blood glucose or insulin levels, and pancreatic architecture and insulin immunoreactivity were similar to control, yet their brains were atrophied and had striking evidence of neurodegeneration with cell loss, gliosis, and increased immunoreactivity for p53, activated GSK-3β, phospho-tau, ubiquitin, and APP-Aβ. 28,29 Moreover, quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) studies demonstrated that the ic-STZ-treated brains had significantly reduced expression of genes corresponding to neurons (Hu), oligodendroglia [myelin-associated glycoprotein-1 (MAG-1)], and ChAT and to increased expression of genes encoding GFAP, microglia-specific proteins [allograft inflammatory factor-1 (AiF-1)]), acetylcholinesterase (AChE), tau, and APP.28,29 Increased p53 and decreased Hu and MAG-1 expression in ic-STZtreated brains suggest that neuronal and oligodendroglial cell loss and cerebral atrophy were mediated by apoptosis. These findings correspond well with previous studies demonstrating increased expression of various proapoptosis molecules, including p53,105,106 colocalization of increased p53 immunoreactivity in neurons and white matter glia, and reduced levels of Hu and MAG-1 mRNA in human brains with AD. Loss of oligodendroglia could contribute to the early white matter degeneration107 and synaptic disconnection108–111 in AD.
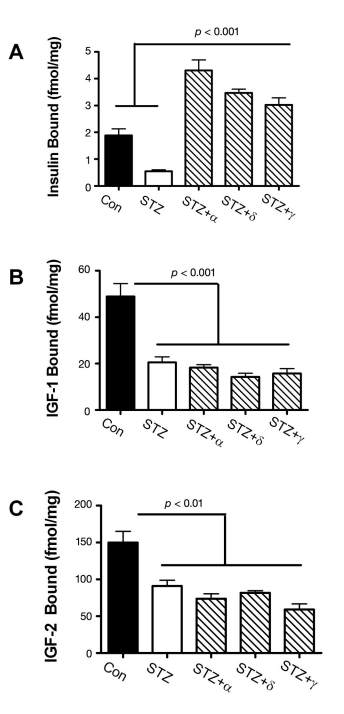
The previously mentioned adverse effects of ic-STZ were associated with reduced expression of genes encoding insulin, IGF-2, insulin receptor, IGF-1 receptor, and IRS-1 and reduced ligand binding to the insulin and IGF-2 receptors (Figure 3). Note that most of these effects were also detected in brains with sporadic AD5 and were found to increase with disease progression.10 The reduced levels of IRS-1 mRNA observed in both AD and rats treated with ic-STZ were reminiscent of the murine IRS-1 and insulin receptor knock-out models, which exhibit reduced brain and body weights due to impaired insulin stimulated growth and survival signaling.23,112,113
Figure 3. Effects of intracerebral ic-STZ treatment on CNS expression of insulin and IGF (A) genes and (B) receptors and (C) ligand binding to the insulin, IGF-1, or IGF-2 receptors in temporal lobe tissue. Rat pups were given 50 mg/kg ic-STZ or vehicle and sacrificed 14 days later. Temporal lobe mRNA was used to measure gene expression by qRT-PCR, and results were normalized to 18S rRNA measured in the same samples.29 Note axis break in Panel A. (C) Competitive equilibrium binding assays were used to measure specific binding to the insulin, IGF-1, or IGF-2 receptors as described in Figure 2. Graphs depict the mean ± standard error of the mean of results. Data were analyzed statistically using Student’s t-tests. Significant p-values are indicated over the bar graphs.
The combined effects of reduced insulin/ IGF polypeptide gene expression, receptor expression, receptor binding, and IRS expression all point toward failure of insulin/IGF signaling mechanisms in the brain as a major consequence of ic-STZ treatment. Importantly, many molecular abnormalities that characteristically occur in AD and were produced by ic-STZ, including increased GSK-3β activation, increased tau phosphorylation, and decreased neuronal survival, could be mediated by downstream effects of impaired insulin and IGF signaling in the CNS. Again, similar results have been reported by other investigators using this experimental model of neurodegeneration.114–117 Therefore, the ic-STZ experimental animal model recapitulates many of the characteristic features of AD-type neurodegeneration/T3DM. Corresponding with the findings in AD,5 the ic-STZtreated brains had increased levels of activated GSK-3β, phospho-tau, ubiquitin, APP and APP-Aβ and decreased levels of tau protein. These results are consistent with previous studies demonstrating that tau is regulated by insulin/IGF-1 stimulation88,118 and that tau phosphorylation and ubiquitination increase with oxidative stress and activation of GSK-3β. 93 Similarly, APP mRNA increases with oxidative stress and is a feature of sporadic AD.5,10 Increased APP gene expression could account for APP-Aβ accumulation in AD and ic-STZ-treated brains. Potential sources of oxidative stress in AD and the ic-STZ model include (1) mitochondrial dysfunction;6,53,95 (2) microglial cell activation with increased cytokine release; and (3) impaired insulin/IGF signaling through PI3 kinaseAkt, leading to increased levels of GSK-3β activity. A crucial step was to determine whether ic-STZ could cause disturbances in acetylcholine homeostasis and cognitive impairment as they occur in AD. QRT-PCR and immunohistochemistry detected reduced levels of ChAT and increased levels of AChE mRNA and protein in icSTZ-treated brains relative to control brains. Note that energy metabolism leads to production of Acetyl-CoA, which is needed to make acetylcholine. Since the ChAT gene is responsive to insulin and IGF-1 stimulation, deficits in insulin/IGF signaling and energy metabolism push in the direction of cholinergic deficiency mediated by impaired energy metabolism and decreased expression of ChAT, which are key features in AD. In addition, increased levels of AChE expression in the ic-STZ brains could result in increased degradation of acetylcholine thereby exacerbating the acetylcholine deficits caused by reduced ChAT expression. The significance of these results is highlighted by the prominent learning and memory deficits detected in ic-STZ-treated rats.
Type 3 Diabetes May Be Treatable, Preventable, or Curable with Antidiabetes Drugs
The findings that (1) pronounced insulin/IGF deficiency and resistance develop early in the course of AD; (2) insulin/IGF signaling abnormalities progress with severity of neurodegeneration;5,10 and (3) an experimental animal model with features closely mimicking the molecular, biochemical, and neuroanatomical pathologies of AD could be generated by intracerebral delivery of a drug that causes T1DM or T2DM led us to test the hypothesis that AD-type neurodegeneration and cognitive could be reduced or prevented by early treatment with insulin-sensitizer antidiabetes agents such as peroxisome proliferator-activated receptor (PPAR) agonists. Peroxisome proliferator-activated receptor agonists function at the level of the nucleus to activate insulin-responsive genes and signaling mechanisms. PPAR-α, PPAR-δ, and PPAR-γ are all expressed in adult human brains, including AD, but PPAR-δ is the most abundant of the three isoforms.6 The experimental design involved treating rats with ic-STZ, followed by a single intraperitoneal injection of saline, a PPAR-α (GW7647; 25 µg/kg), PPAR-δ (L-160,043; 2 µg/kg), or PPAR-γ (F-L-Leu; 20 µg/kg) activator (CalBiochem, Carlsbad, CA).28 The doses used were considerably lower than those routinely given to treat T2DM. The major effects of the PPAR agonist treatments were to prevent brain atrophy, preserve insulin and IGF-2 receptor bearing CNS neurons, and particularly with regard to the PPAR-δ agonist, prevent ic-STZ-induced deficits in learning and memory.28 Since the ic-STZ-mediated losses of insulin and IGF-expressing cells were not prevented by the PPAR agonist treatments, the PPAR agonists probably functioned by preserving insulin and IGF responsive (receptor-bearing) cells, including neurons and oligodendroglia. In support of this concept was finding that insulin receptor expression and binding were increased by the PPAR agonist treatments (Figure 4). Peroxisome proliferator-activated receptor agonist mediated preservation of insulin/IGF responsive neurons was associated with increased expression of ChAT, which has an important role in cognition, as cholinergic neuron deficits are a fundamental feature of AD.119–122 Importantly, the PPAR-δ agonist mediated increases in insulin binding, and ChAT were associated with significant improvements in learning and spatial memory tasks as demonstrated using Morris water maze tests28
(Figure 5). These effects of the PPAR agonist treatments are consistent with the facts that ChAT expression is regulated by insulin/IGF88,118 and insulin/IGF resistance mediates cognitive impairment in AD. The PPARmediated increases in MAG-1 expression, corresponding to oligodendroglia, were of particular interest because previous research demonstrated that one of the earliest AD lesions was white matter atrophy and degeneration with loss of oligodendroglial cells.107 Within the context of the present discussion, white matter atrophy in AD can now be interpreted as a manifestation of CNS insulin/IGF resistance since oligodendroglia require intact insulin/IGF signaling mechanisms for survival and function, including myelin synthesis.123,124 Besides preserving insulin and IGF receptor-bearing CNS cells and signaling mechanisms germane to survival, energy metabolism, and neurotransmitter functions, the PPAR agonists rescued the ic-STZ model by lowering critical ADassociated indices of oxidative stress, including microglial and astrocyte activation, p53, nitric oxide synthase and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase gene expression, lipid peroxidation, DNA damage, APP expression, and tau phosphorylation.6,28,29,91,92,125,126
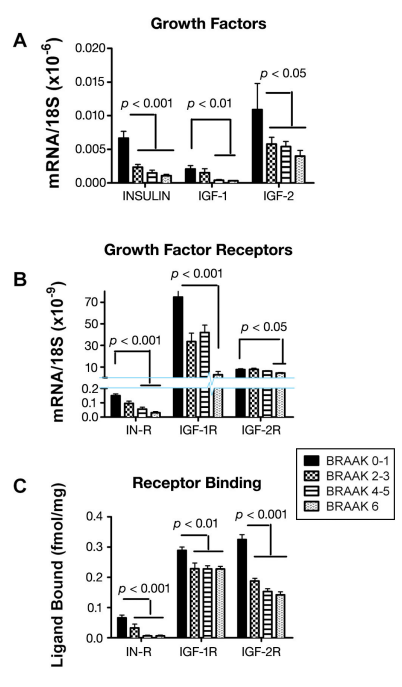
Figure 4. Treatment with PPAR agonists restores brain insulin receptor binding in ic-STZ-treated rats.28 Long Evans rat pups were treated with 50 mg/kg ic-STZ or vehicle and sacrificed 30 days later to examine brains for insulin and IGF polypeptide and receptor gene expression and insulin and IGF receptor binding. Temporal lobe membrane protein extracts were used in competitive equilibrium binding assays to measure specific binding to the (A) insulin, (B) IGF-1, or (C) IGF-2 receptors as described in Figure 2. Graphs depict the mean ± standard error of the mean of results. Data were analyzed using ANOVA with the Tukey–Kramer post hoc significance test. Significant p-values are shown within each panel.
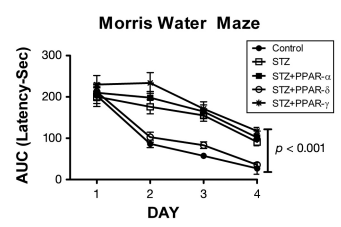
Figure 5. Peroxisome proliferator-activated receptor-δ agonist treatment preserves visual-spatial learning and memory in ic-STZ-treated rats.28 Long Evans rat pups were treated with 50 mg/kg ic-STZ or vehicle, followed by a single intraperitoneal injection of a PPAR-α (GW7647; 25 µg/kg), PPAR-δ (L-160,043; 2 µg/kg), or PPAR-γ (F-L-Leu; 20 µg/kg) agonist (n = 8 rats per group). Four weeks later, the rats were subjected to Morris water maze testing, in which the latency required to locate the hidden platform was measured for 3 independent trials on 4 consecutive days. Area under the curve (AUC) was computed for the 3 daily trials. Graphs depict the mean AUC ± standard error of the mean for latency (seconds) in each group. Data were analyzed using ANOVA with the Tukey–Kramer post hoc significance test. Performance in the control and ic-STZ + PPAR-δ groups were similar, and on Days 2, 3, and 4, their mean latencies required to locate the hidden platform were significantly shorter than in the other 3 groups.
Altogether, the results from these studies provide strong evidence in support of the hypothesis that AD represents a form of diabetes mellitus that selectively afflicts the brain. Positive data stemmed from (1) direct analysis of postmortem human brains with documented AD; (2) an experimental animal model in which brain diabetes with cognitive impairment and molecular and pathological features that mimic AD was produced by intracerebral administration of a drug that is commonly used to produce T1DM or T2DM; and (3) a study showing that PPAR agonists, which are used to treat T2DM, prevent many of the AD-associated neurodegenerative effects of ic-STZ. The data are supported by abundant in vitro experiments that demonstrated essentially the same or similar effects of STZ or oxidative stress treatments of neuronal cells. The human and experimental animal model studies also showed that CNS impairments in insulin/IGF signaling mechanisms can occur in the absence of T1DM or T2DM. Finally, we demonstrated that although obesity with T2DM causes brain insulin resistance with some features of AD-type neurodegeneration, the effects are relatively modest, not associated with significant histopathological lesions, and lack most of the critical abnormalities that typify AD. Therefore, T2DM was deemed not sufficient to cause AD, although it could possibly serve as a cofactor in its pathogenesis or progression. Altogether, the data provide strong evidence that AD is intrinsically a neuroendocrine disease caused by selective impairments in insulin and IGF signaling mechanisms, including deficiencies in local insulin and IGF production. At the same time, it is essential to recognize that T2DM and T3DM are not solely the end results of insulin/IGF resistance and/or deficiency, because these syndromes are unequivocally accompanied by significant activation of inflammatory mediators, oxidative stress, DNA damage, and mitochondrial dysfunction, which contribute to the degenerative cascade by exacerbating insulin/ IGF resistance. Referring to AD as T3DM is justified, because the fundamental molecular and biochemical abnormalities overlap with T1DM and T2DM rather than mimic the effects of either one. Some of the most relevant data supporting this concept have emerged from clinical studies demonstrating cognitive improvement and/or stabilization of cognitive impairment in subjects with early AD following treatment with intranasal insulin or a PPAR agonist.58,60,127–130
Conclusions
We need your consent to load the translations
We use a third-party service to translate the website content that may collect data about your activity. Please review the details in the privacy policy and accept the service to view the translations.